质量安全与营养品质评价岗位
李雪 张玉 王强
花色苷类化合物是葡萄中重要的酚类物质,主要存在于葡萄的外果皮和种子中,对葡萄和葡萄酒的品质具有重要作用,同时花色苷还具有抗氧化、抗肿瘤、抗过敏、保护胃黏膜、预防冠心病等多种保健功能。花色苷是红葡萄和红葡萄酒颜色的主要来源,以C6-C3-C6为骨架,C环上C3位羟基经过糖苷化反应形成,花色苷可进一步被酰基化修饰,生成乙酰化、香豆酰化和咖啡酰化的花色苷,其种类极其丰富。
目前花色苷检测前处理方法主要采用传统的有机溶剂浸提法,该法存在效率低、耗时长、萃取有机溶剂消耗量大等问题。随着检测技术的发展,在检测前处理中,运用更有效、更环保的提取方法越来越受到人们重视。天然深层共晶溶剂(NADES)是由形成分子内氢键的两个或三个成分组成的混合物,是一种潜在的绿色溶剂。目前葡萄中花色苷的定量方法主要以外标法定量,但由于其花色苷种类繁多,部分对照品难以获得,同时价格较高,大大提高检测成本。因此,本研究采用天然深层共晶溶剂对样品进行提取,选择含量相对较高、性质稳定、较易获得的花色苷化合物为内参物,建立超高效液相色谱-高分辨质谱结合一测多评法同时定性、定量分析葡萄中多种花色苷成分的方法,旨在为葡萄的品质评价及开发利用提供依据。
1 仪器与材料
1.1 仪器
Waters Acquity UPLC液相色谱串联Waters Synapt G2S四级杆飞行时间高分辨质谱仪(美国Waters公司);RV10-D-V型旋转蒸发器:艾卡(广州)仪器设备有限公司。
1.2 材料与试剂
甲基花青素-3-O-葡萄糖苷、锦葵色素-3-O-葡萄糖苷、飞燕草色素-3-O-葡萄糖苷、矢车菊素-3-O-葡萄糖苷、天竺葵色素-3-O-葡萄糖苷、甲基花翠素-3-O-葡萄糖苷、锦葵色素-3,5-O-双葡萄糖苷、芍药素-3,5 -O-双葡萄糖苷、天竺葵素-3,5 -O-双葡萄糖苷、花青素-3,5 -O-双葡萄糖苷标准品(纯度>99%):阿拉丁试剂公司;甲醇为国产分析纯;乙腈:色谱纯,德国默克公司;超纯水:Millipore纯水仪制备。
葡萄样品:夏黑、赤霞珠、马瑟兰葡萄果实,采后放入-80℃超低温冰箱备用。
1.3 提取方法
1.3.1常规溶剂提取
葡萄于液氮中研磨至粉,准确称取1 g葡萄粉加入40 mL 1%盐酸甲醇溶液,于30℃避光提取2 h,离心(4℃,8000 r/min,5 min),重复提取3次,合并提取液,35℃旋转蒸发至干,残留物用甲醇定容至5 mL,待测。
1.3.2天 然深层共晶溶剂制备
NADES选择的第一步是根据目标化合物的性质,选择氢键供体和受体。花青素是一种高极性化合物,在水中溶于非极性溶剂,其化学形式和稳定性取决于pH值。花青素普遍呈黄藻阳离子形式,在酸性时稳定,在pH>7时降解。根据特性制备了6种不同的NADESs。该方法包括在80℃的搅拌下,以适当的摩尔比混合化学成分,直到形成透明的无色液体(表1)。每种混合物的pH使用装有电极探头的Crison2002pH计进行测量,室温下保存待测。
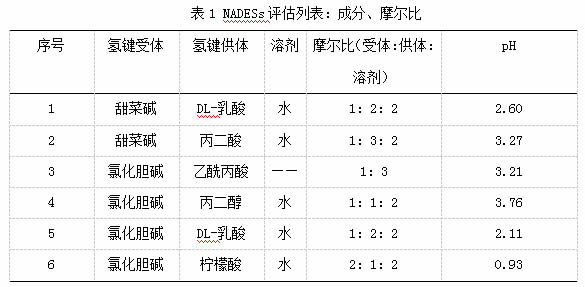
1.3.3 NADESs法提取葡萄皮花色苷
准确称取葡萄粉末1 g加入10mL制好的不同深层共晶溶剂,于50℃超声提取30 min,离心(4℃,8000 r/min,15 min),取上清液于测定。
1.4 花色苷含量的测定
1.4.1 总花色苷的测定
采用pH示差法测定样品中花色苷总含量。
1.4.2 液质分析
仪器条件
色谱条件:色谱柱:Waters ACQUITY UPLC BEHC18 柱(2.1 mm×100 mm,1.7 μm)。柱温:35℃;样品室温度:15℃。流动相:A为1%甲酸水(V/V),B为乙腈。梯度洗脱:0-1 min,95.0-90.0% A;1-6 min,90.0-90.0% A;6-9 min,90.0-75.0% A;9-11 min,75.0-5.0% A;11-13 min,5.0-5.0% A;13-13.5 min,5.0-95.0% A;13.5-18 min,95.0-95.0% A。流速为0.3 mL/min。
质谱条件:ESI电喷雾离子源(美国Waters)。正离子模式;毛细管电压:3.0 kV;锥孔电压,35 V;离子源温度,80℃;脱溶剂气温度,300℃;锥孔气流量,50 L/h ;脱溶剂气流量,600 L/h。质谱采集范围:m/z 100-1000。校正曲线:甲酸钠,分辨率模式。
对照品溶液的制备
分别精密称取甲基花青素-3-O-葡萄糖苷、锦葵色素-3-O-葡萄糖苷、飞燕草色素-3-O-葡萄糖苷、矢车菊素-3-O-葡萄糖苷、天竺葵色素-3-O-葡萄糖苷、甲基花翠素-3-O-葡萄糖苷、锦葵色素-3,5-O-双葡萄糖苷、芍药素-3,5 -O-双葡萄糖苷、天竺葵素-3,5 -O-双葡萄糖苷、花青素-3,5 -O-双葡萄糖苷对照品适量,加甲醇制成浓度分别为10、9.75、19.8、30.0、9.975、19.8、69.9、14.85、30.0、50.0 μg/mL的混合对照品溶液。
1.4.3 方法学考察
1.4.3.1 线性关系考察
精密吸取混合对照品溶液适量分别稀释2、4、10、20、40倍,按“1.4.2”项下仪器条件进样测定。以对照品浓度为横坐标,峰面积为纵坐标,进行线性回归。
1.4.3.2 精密度实验
精密吸取混合对照品溶液适量,按“1.4.2”中仪器条件,同一天连续进样测定6次,并连续3天进样,计算日内精密度及日间精密度。
1.4.3.3 重复性实验
精密称取同一批样品6份,按“1.4.2”中方法制备供试溶液,并进样测定,计算花色苷化合物的含量及RSD值。
1.4.3.4 回收率实验
称取葡萄样品,加入10种标准品,按照1.3.3节方法制备花色苷提取物,再按照1.4.2节色谱条件进样,并计算10种化合物的加标回收率。
1.5 相对校正因子(F)的计算
相对校正因子(F)的测定依据下列公式:
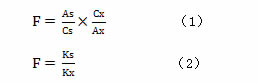
式中As为内参物的峰面积,Cs为内参物的浓度,Ax为化合物X的峰面积,Cx为化合物X的浓度;Ks为内参物的线性回归方程斜率,Kx为化合物X的线性回归方程斜率。公式(1)为校正因子多点计算方法,公式(2)为校正因子斜率计算方法。
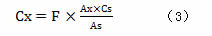
公式(3)为以校正因子计算化析物X浓度的方法。
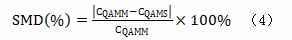
一测多评方法的准确性以公式(4)计算的相对标准偏差(SMD%)进行评价,CQAMM为以外标法计算的化合物浓度,CQAMS为以一测多评法计算的化合物浓度。
1.6 数据分析
本研究获得的数据采用3次重复实验的平均值和相对标准偏差(RSD)表示。利用GraphPad Prism5软件进行数据统计分析。
2 结果与讨论
2.1 不同提取方式筛选
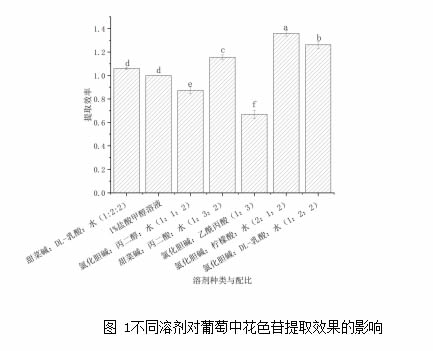
采用常规提取方法和不同深层共晶溶剂提取,测定花色苷的总量,以常规酸化甲醇提取率为1,其他提取率以相对酸化甲醇提取率比例表示,结果显示氯化胆碱:柠檬酸:水(2:1:2)的提取率最高,其次为氯化胆碱:DL-乳酸:水(1:2:2)并优于常规酸化甲醇提取法(图1)。
2.2 液相色谱质谱条件优化
本研究首先对花色苷组分的液相色谱-高分辨质谱条件进行了优化。对于液相色谱条件,首先对流动相进行了考察,分别以纯水、0.1%甲酸-水、0.5%甲酸-水及1.0%甲酸-水作为水相,对比分离结果,在1.0%甲酸-水中获得更好的峰形及灵敏度,因此选择%甲酸-水作为水相;本研究还对比了Waters ACQUITY UPLC BEH C18 (100 mm ×2.1 mm, 1.7 µm)及Waters ACQUITY UPLC RP18两种色谱柱的分离效果,后者所需时间长,且分离度较低,因此选择BEH 18柱用于色谱分离。对于质谱条件,主要考察了待测成分在正负谱中的信号响应,结果显示,正离子模式能提供更多的化合物种类信息,因此采用正离子模式进行分析;其它质谱参数通过仪器自动优化获得。优化后的液相色谱质谱条件见1.4.2节。
2.3 葡萄中花色苷分析方法验证
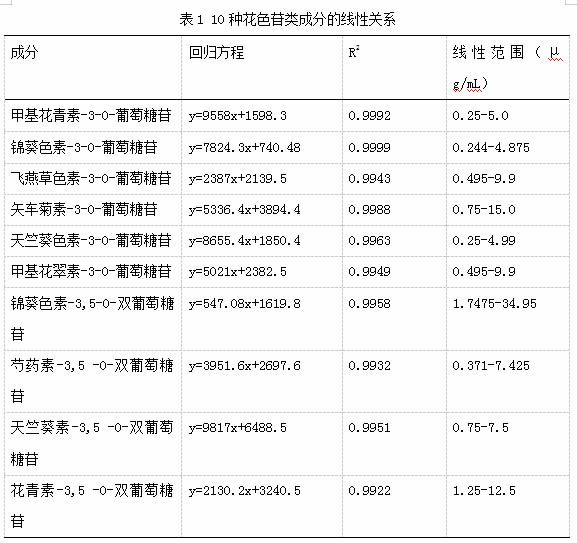
如表1所示,10种化合物的峰面积和质量浓度之间均呈良好的线性关系,相关系数(R2)在0.9932~0.9999之间,检出限及定量限分别按照信噪比3、10计算,检出限范围为0.08~0.58 μg/mL,定量限为0.24~1.74 μg/mL。将10种花色苷在同一天连续进样6次的峰面积的RSD作为日内精密度,分别为3.45%、4.00%、1.33%、2.62%、3.99%、3.08%、4.09%、4.45%、0.58%、3.31%。并将其连续进样三天的峰面积的RSD作为日间精密度,分别为2.35%、2.28%、0.79%、1.65%、0.61%、1.71%、3.72%、4.23%、0.80%、1.87%。结果表明仪器精密度良好。采用同一批样品重复进样,计算重复性,RSD%在1.22%~2.43%之间,表明本方法准确可靠。回收率结果表明,10种化合物的加标回收率在93.42%~119.86%之间,RSD≤3.12%,表明实验方法有较好的回收率,回收结果准确可靠。
2.4 相对校正因子的测定
目前,一测多评方法主要与HPLC-UV及HPLC-QqQ-MS相结合用于不同化合物的定量测定,但是以上方法对于复杂基质化合物的同时定性、定量具有一定的局限性,而高效液相色谱结合高分辨质谱在准确定性的基础上,降低基质干扰而准确定量具有优势。因此,本研究采用超高效液相色谱-高分辨质谱结合一测多评理论建立葡萄中花色苷的快速、精准定量方法。
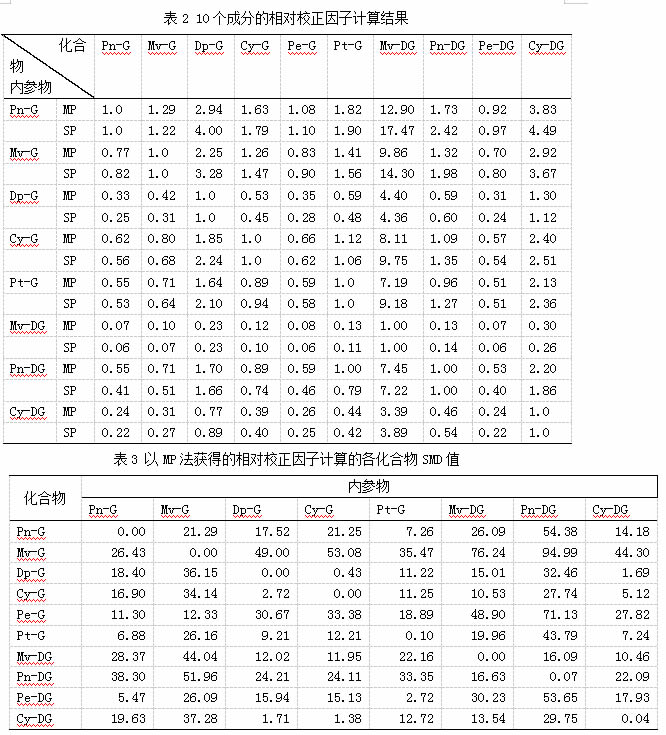
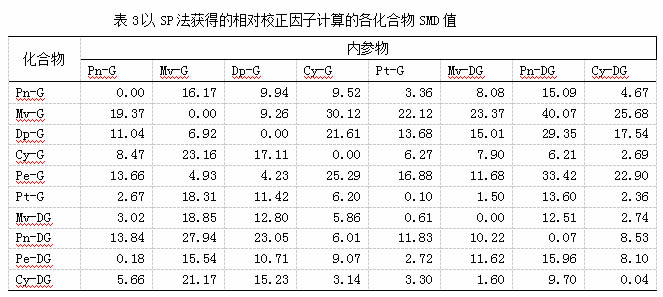
一测多评法中相对校正因子的计算是影响方法准确性的关键因素,而葡萄中花色苷类化合物复杂多样,内参物的选择对相对校正因子的计算具有较大影响。本研究中以不同花色苷化合物作为内参物,采用多点法(MP)及斜率法(SP)分别计算各化合物相对校正因子,其中MP法是以各化合物在不同浓度下的F值的平均值做为相对校正因子,结果见表2。在以MP法计算校正因子时,选择不同花色苷化合物作为内参物计算得到的校正因子相对标准偏差(RSD%)差异较大1.29%~26.04%,其中内参物为甲基花青素-3-O-葡萄糖苷、矢车菊素-3-O-葡萄糖苷、锦葵色素-3-O-葡萄糖苷时,除了花青素-3,5 -O-双葡萄糖苷RSD值在10%以上外,其它化合物RSD值均低于10%,表明相对校正因子的计算较准确。但MP与SP法计算的相对校正因子比较结果可见,二者存在一定的差异。为了验证及比较两种计算方法的准确性,进一步采用SMD值进行结果评估,结果见图1。由图可见,以SP法获得的相对校正因子计算的化合物浓度与外标法所得结果差异更小,其中甲基花青素-3-O-葡萄糖苷为内参物时,SMD值在2.67%~19.37%,除了锦葵色素-3-O-葡萄糖苷(19.37%)外,均在14%以下,表明其化合物计算浓度与外标法基本一致,具有较高的准确性。
2.5 葡萄样品中花色苷类化合物的分析
本研究在葡萄样品中检测到12种花色苷类化合物,包括单糖苷、双糖苷、香豆酰化及咖啡酰化的花色苷类化合物。其中夏黑品种中花色苷种类最多,检测到5种单糖苷、2种双糖苷及4种酰化糖苷类花色苷,而5种单糖苷飞燕草色素-3-O-葡萄糖苷、矢车菊素-3-O-葡萄糖苷、甲基花翠素-3-O-葡萄糖苷、甲基花青素-3-O-葡萄糖苷、锦葵色素-3-O-葡萄糖苷及锦葵色素-3-O-(6-O-香豆酰化)-葡萄糖苷在所有样品中均有检出。采用本文建立方法对样品中花色苷含量分析表明(表3),不同样品中花色苷类化合物含量差异较大,飞燕草色素-3-O-葡萄糖苷、矢车菊素-3-O-葡萄糖苷、甲基花翠素-3-O-葡萄糖苷、甲基花青素-3-O-葡萄糖苷、锦葵色素-3-O-葡萄糖苷在不同样品中的含量范围分别为10.30~493.02μg/g、0.93~67.45 μg/g、13.83~192.11 μg/g、1.09~259.46 μg/g、20.39~1000.58 μg/g 其中马瑟兰中锦葵色素-3-O-葡萄糖苷含量最高(1000.58 μg/g),而赤霞珠中飞燕草色素-3-O-葡萄糖苷含量较高。
3 结论
本实验首次建立了基于深共晶溶剂结合一测多评法测定葡萄中多种花色苷的检测技术。方法通过氢键供体和受体的组成及合成比例筛选得到最佳深共晶溶剂提取剂氯化胆碱:柠檬酸:水(2:1:2),经超声提取后,用超高效液相色谱-质谱分析,以甲基花青素-3-O-葡萄糖苷为内参物,建立其与其它9种化合物的相对校正因子,采用一测多评法进行定量计算,结果表明,一测多评法的相对校正因子重现性良好,与外标法测得值无显著差异。该方法简便、准确,可用于葡萄中花色苷的含量测定。