病毒病防控岗位
范旭东 董雅凤 张尊平 任芳 胡国君
国际病毒分类委员会将芜菁黄花叶病毒科划分为3个属,分别为芜菁黄花叶病毒属(Tymovirus)、玉米细线病毒属(Marafivirus)和葡萄斑点病毒属(Maculavirus)。这3个属的RNA病毒成员一般具有长度6.0-7.5kb的基因组。侵染葡萄的此类型病毒已报道5种,即葡萄斑点病毒(GFkV)、葡萄红球病毒(GRGV)、葡萄西拉病毒1(GSyV-1)、葡萄星状花叶相关病毒(GAMaV)和沙地葡萄叶脉羽化病毒(GRVFV)。其中,GFkV和GRGV属于Maculavirus,GSyV1、GAMaV和GRVFV属于Marafivirus。已有研究表明GFkV、GRGV、GAMAV和GRVFV与葡萄斑点复合病相关。葡萄斑点复合病是一种分布于世界各地的主要葡萄病毒病之一,由几种病害类型组成,包括葡萄斑点(FK)、葡萄星状花叶(AM)、沙地葡萄坏死和沙地葡萄羽化等。AM是最早在美国加利福尼亚发现的葡萄斑点复合病类型,它能通过嫁接进行传播,其症状特征是在几个葡萄品种叶片上有半透明或褪绿的星状斑点,也导致沙地葡萄的初级和次生叶脉产生脉明症状。病毒病防控岗位于2014年在大连市的‘巨玫瑰’葡萄上发现了明显的叶脉褪绿症状,这些病症与AM极为相似,因此当时推测可能的病原为GAMaV,但在该样本中未检测到。2017年,为了确定感病葡萄样本中可能存在的病毒种类,采用了小RNA测序(sRNA-seq)和RNA测序(RNA-seq)对其进行了病毒鉴定,发现了一种新的葡萄病毒,命名为葡萄玉米细线病毒属相关病毒(grapevine associated-marafivirus,GaMV),并对我国葡萄GaMV进行了检测和基因序列分析。
1 材料和方法
1.1 试验材料
2014年,从大连的一个葡萄园采集到一株具有明脉症状的‘巨玫瑰’葡萄(图1a,b)。从感染的葡萄上繁殖的插条扦插成活后,其叶片也表现出脉明症状(图1c, d和e)。2017年春季,采集病叶在液氮中快速冷冻,通过干冰运送至百迈克生物科技公司。
1.2 研究方法
1.2.1 高通量测序和生物信息学分析
提取叶片总RNA,并构建sRNAs cDNA文库,采用Illumina HiSeq™2000系统进行sRNA-Seq。从原始读数中去除<18nt或>30nt的序列、低质量的、包含PolyA和N的序列,以获得过滤后的数据。通过使用VirusDetect分析小RNA数据,确定了潜在病毒的序列。对于RNA-seq,使用EpicentreRibo-Zero rRNA Removal Kit(Epicentre,Madison,WI,USA)从总RNA提取物中去除核糖体RNA。然后,使用TruSeq RNA Sample Prep Kit(Illumina,San Diego,CA,USA)使用核糖体RNA缺失RNA样品构建cDNA文库,该cDNA文库在Illumina HiSeq 4000平台上以成对的150bp测序。比对上的葡萄基因组(PN40024组装12X)的序列,使用hisat软件移除。未比对上的序列通过VirusDetect软件进行拼接和BLAST分析。

1.2.2 GaMV基因组扩增及序列分析
根据contigs序列设计了5对引物,用于扩增GaMV基因组序列。回收、纯化PCR片段,将其克隆到零背景pTOPO-Blunt载体(Aidlab,中国,北京)。每个PCR产物至少3个阳性克隆送上海生物工程技术公司进行了测序。采用SMARTer®RACE 5’/3’Kit (TaKaRa)试剂盒对GaMV的5’和3’UTR进行扩增。
1.2.3 GaMV不同分离物序列分析
采用NCBI在线的ORF Finder软件分析序列潜在的ORFs。保守结构域通过CD工具来分析。Emaraviruses的核苷酸和氨基酸的序列比对通过ClustalW软件进行。采用MEGA7.0对RNA1-4编码的蛋白进行进化树分析。
1.2.4 嫁接传染实验
2019年7月,从感染GaMV的葡萄上采芽嫁接到2年生的Beta葡萄幼苗上,设5个重复。用于嫁接的贝达苗经检测,不携带GAMV和中国报道的其他主要病毒,且无病毒病症状。接种后3、6和12个月,监测嫁接葡萄的症状发生情况。提取被嫁接苗的RNA,采用两对引物(RP1a/1b和CP1a/1b)扩增GaMV的RdRp和CP基因序列,以确定样品是否被传上GaMV。
1.2.5 田间样品GaMV检测
为明确我国GaMV的流行情况,从我国21个省市随机采集了71个品种516株葡萄,采用RP1a/1b和CP1a/1b引物进行GaMV的检测。
2 结果与分析
2.1 高通量测序结果分析
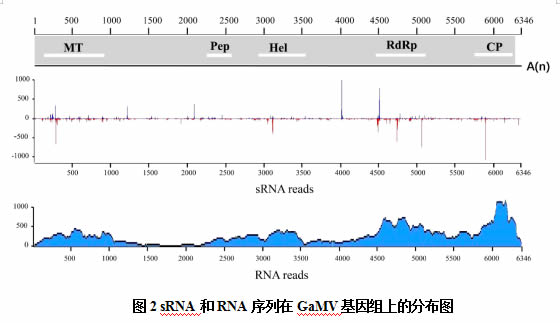
对巨玫瑰葡萄病叶中的sRNA和RNA进行测序,过滤得到18,599,455和602,231,929条序列。采用VirusDetect软件对序列进行分析,鉴定出样品中潜在的病毒序列。sRNA-Seq序列显示17个长度为55~286nt的contigs与GAMaV(AOX24075)的多聚蛋白同源,覆盖率为26.6%;RNA-Seq数据显示有14个长度为206~4866nt的重叠群与燕麦蓝矮病毒(AAC57874)的多聚蛋白同源,覆盖率为94.6%。这些结果表明在巨玫瑰葡萄样品中存在一种潜在的Marafivirus,我们暂定将其命名为葡萄相关Marafivirus(GaMV)。来自sRNA-seq和RNA-seq数据的50,789个sRNA和12,937个RNA能够比对上GaMV(图2)。
2.2 GaMV基因组序列及分析
根据contigs序列设计引物,RT-PCR扩增了GaMV JMG分离物的5个重叠片段,并通过重叠的公共序列(一般>100bp)组装成一个连续的序列。此外,我们还利用3’ cDNA末端快速扩增(RACE)获得了3’个非翻译区(UTR)的完整序列,并通过5’ RACE获得了5’端的部分序列。最终,获得了GAMV的近全基因组(GenBank登录号:MZ422607),共6346bp,不包括PolyA尾部序列。为明确GaMV与Tymoviridae科其它成员之间的关系,构建了一个系统发育树。进化树分析使用了所有已确认的Marafivirus,Maculavirus和Tymovirus成员。分析结果显示,GaMV与其它marafivirus成员聚在一起(图3)。

2.3 GaMV嫁接传染特性
所有嫁接的‘贝达’葡萄都表现出明显的脉明和褪绿斑点症状(图4 a-c)。嫁接12个月后,5株‘贝达’葡萄用两对特异的PCR引物均检测出GaMV(图4 d)。未嫁接植株GaMV检测结果呈阴性,且未表现症状。这些数据表明,GAMV可以通过嫁接传染。
2.4 田间样品GaMV检测
采用引物CP1a/1b和RP1a/1b对516份样品进行RT-PCR检测,阳性样品分别为23份(4.46%)和12份(2.33%),表明前一对引物比后一对引物检测效果好。合计检测到阳性样品26个,占总样品的5.04%,其中,北京1个、辽宁21个、宁夏2个、山东1个、四川1个。
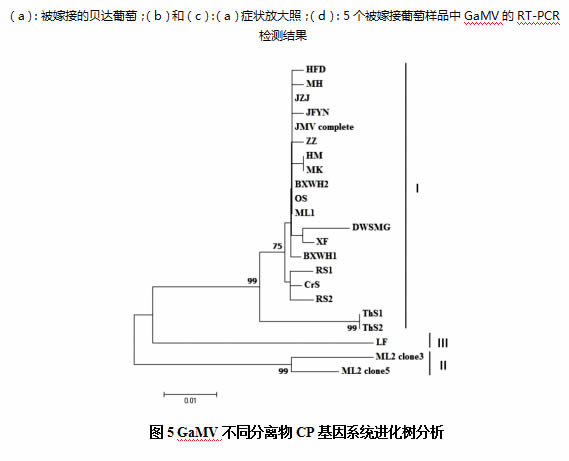
2.5 不同分离物GaMV CP基因序列同源性及进化树分析
研究获得了20个GAMV分离物的CP序列,分析了GAMV分离物之间的遗传多样性。这些分离株的序列已登录GenBank。除ML2外,其余分离株在的3个初始测序克隆的核苷酸同源性均>99.0%的。因此,对菌株ML2进行了更多的克隆测序,并鉴定出以ML2克隆3和ML2克隆5为代表的两种变异类型。GAMV分离物的CP在核苷酸和氨基酸水平上的同源性分别为91.7-100%和96.7-100%。基于CP的系统发育树揭示了三个定义明确的类群的存在。主要菌株属于群I,而菌株ML2和LF分别属于群II和群III(图5)。